Coronaviruses 101: Focus on Molecular Virology
May 03, 2020Hello, my name is Britt Glaunsinger, I am a virologist and professor at the University of California, Berkeley and a researcher at the Howard Hughes Medical Institute. And what I'm going to do is present a lecture on the fundamental
molecular
virology
ofcoronaviruses
. These are viruses that have been circulating in the human population and animals for a long time. We know of seven humancoronaviruses
. These are present in two of the four known genera of coronaviruses, alpha coronaviruses and beta coronaviruses. The four circulating strains of human coronavirus are shown here in a red box. There are two that we have known about for a long time called 229E and OC43.
These, along with the other two, NL63 and HKU1, which were actually discovered more recently after the SARS epidemic, but are probably also circulating in the human population and have not emerged recently. These four circulating viruses are the causes, some of the causes of the common cold. Probably 10 to 15 percent of common colds are caused by these viruses. We also know of three coronaviruses that have recently emerged in the human population through species jumping or zoonotic transfer and these, of course, are the original SARS coronavirus, the MERS coronavirus, and the newly emerged caronavirus 2, which is the cause of COVID. -19.

More Interesting Facts About,
coronaviruses 101 focus on molecular virology...
Each of these, like other alpha and beta coronaviruses, is thought to have a common ancestor in bat viruses. And this is different from the gamma and delta coronavirus genus, which have common ancestors in birds. The SARS coronavirus and the MERS coronavirus, as I mentioned, probably came from bats. But it is thought that, rather than jumping directly to humans from bats, they first entered the human population through one or more intermediate hosts, animal hosts. For SARS, the intermediate, or at least the primary, animal host is thought to be civet cats and for the MERS coronavirus, the intermediate host is likely to be dromedaries in these intermediate hosts.

The virus likely jumped from bats to these hosts and then underwent a few rounds of replication in these hosts and, in doing so, likely acquired mutations that allowed the virus to spread more easily to the human population. Now, for the purposes of transmission, we don't know what the other intermediate hosts might be, and in particular, we don't know what the intermediate hosts are, if any, for SARS coronavirus 2, the current pandemic strain. It is possible that it came directly from bats. It is also possible that it passed through one or more animals before jumping to humans. I should mention that these are certainly not the only coronaviruses in bats.

More than 500 coronaviruses have been identified in bats in China and estimates of the diversity of coronaviruses from unknown bats reach into the thousands, indicating that they are likely severely undersampled in the bat population. And I think this is also important because it suggests that this current pandemic strain of coronavirus 2 is unlikely to be the last one we'll see if coronaviruses, in particular, we've already had three zoonotic jumps of highly pathogenic coronaviruses in the human population in less than twenty. years and with the enormous diversity of coronaviruses that probably circulate in bats, I think many scientists and virologists are quite concerned that they will continue to jump into the future as well.
And so it is very worthy of continued study even after this current pandemic is over. It is worth thinking about comparing the other two highly pathogenic zoonotic coronaviruses, SARS and MERS, with the current pandemic. Thus, SARS, which emerged at the end of 2002 and caused just over 8,000 cases and 774 deaths worldwide. This was an epidemic that lasted about a year. It was mostly brought under control in 2003; the latest cases were seen as some kind of laboratory outbreak related to 2004. That epidemic is now over. MERS, on the other hand, has caused a lower number of 2,521 cases to date with 866 deaths in total.
This is actually the most pathogenic virus and the one with the highest mortality rate, around 34 percent. Unlike SARS, MERS infections still occur periodically, and this is probably not due to circulation among the human population. This virus does not transmit particularly well from person to person, but it is believed that these new infections occur due to occasional and recurrent infections from dromedaries to the human population. So why was the SARS epidemic able to be controlled in about a year, while we are clearly far from controlling the current coronavirus 2 pandemic? There are several ideas I have heard discussed about this and I just want to mention three here: Comparing SARS to the current COVID-19 pandemic.
First, the spill reservoir was known, as I mentioned, for the SARS coronavirus. It is mainly the civet cat, and then these animals could be used to try to break the chain and prevent further transmission from these animals to the human population. For Cov-2, as I mentioned, a reservoir spill is not known. Secondly, in the case of SARS Cov-1, most of the human transmission occurred in a hospital environment and, in fact, these hospital environments were centers of transmission of that epidemic, so once it was recognized This and the risk to the medical community was recognized, the staff was able to implement barrier nursing enabled in order to stop the transmission of said virus.
Unlike Cov-2, which has not only spread in a hospital setting but in fact there is widespread community transmission of this virus. And finally, in the case of SARS Cov-1, individuals infected with this virus tended not to transmit until probably 24 to 36 hours after the onset of symptoms and overall, as far as we know, there was a lack of cases asymptomatic. So this is really important from a contact tracing perspective and the ability to stop or inhibit the spread of the virus within the population through effective contact tracing and other public health measures. Unfortunately for Cov-2, the situation is very different, as there are possible and probably abundant asymptomatic cases and further testing will be needed to confirm that mild cases are certainly abundant, which are fueling transmission in the human population.
So, for these reasons and probably other reasons that have to do with
molecular
virology
and epidemiology, the pandemic we are experiencing now is very different from the one we saw in 2003 with SARS and as of this morning it is reaching almost 400,000 confirmed global cases total. . This likely does not reflect the actual number of cases. These are only the cases confirmed by testing, but as we know, there are currently limitations in testing, so the actual number of suspected cases is believed to be much higher. And as of this morning, March 24, 2020, we were reaching over 17,000 deaths, seventeen thousand two hundred and fifty-two deaths worldwide.As you can see from this graph, unfortunately most Western countries are on a very significant coronavirus trajectory, which is basically exponential growth. While some countries have been able to slow growth and limit dispersion, this is largely not the case for the UK, Europe and the US in particular, which show a graph showing clear exponential dispersion, so I think We can anticipate that the number of cases will continue to grow exponentially in the future. All right, we've heard a lot in the news and from experts about the epidemiology of this virus, about the spread of this virus, transmission and control, so that will not be the
focus
of this conference today. .Instead, what I want to do is delve into the molecular virology of how this virus enters and replicates within cells to amplify itself. That is why I have divided the conference into four parts. In the first part, I am going to discuss how the virus can enter cells through interactions with the spike protein and host receptors. Then I'm going to spend time talking about once it deposits its genome into cells, how the virus replicates that genome and expresses its genes. There are some very unusual and interesting features of coronavirus biology in this section. I will then move on to discuss some of the notable cellular biological changes that occur in an infected cell.
In particular, those that involve membranes and the formation of what are called replication and transcription complexes during coronavirus replication. And then at the end, I'm going to spend the last few minutes talking about the immune interactions that this virus has, particularly with the innate immune system, as those are likely drivers of the pathogenesis of these viruses in animal hosts and in humans. Okay, starting with the structure of the viral particle and its entry, we know that viral corona particles are pleomorphic, meaning they don't actually have a defined structure. They have been examined using cryo-electron tomography to confirm this, and they also have what is called a helical nucleic acid.
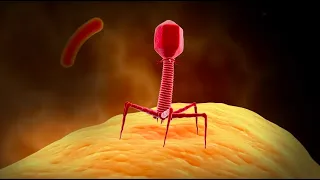
So looking at the structure of the virus, I'm showing you here on the left, the nucleocapsid shown in the center in brown, basically refers to the genome, which is a huge 30 kilobases for a huge RNA virus genome. Huge genome of 30 kilobases of RNA. that is, positive sense or plus sense RNA. When we say positive sense, it means that the cell's ribosomes can read it directly. That genome is coated with a protein called a nucleocapsid protein that forms a kind of helical nucleocapsid. That nucleocapsid-protected genome is basically enclosed in a lipid envelope that is derived from the host cell.
Many viruses have a lipid envelope. In all cases, these lipids are taken from the host. No virus is capable of producing its own lipids, but many viruses use and steal host lipids for replication and sometimes morphogenesis. And that is the case with coronaviruses, where you can see that there is a lipid envelope, which is studded with a number of viral proteins, the most prominent of which is the spike protein shown in blue. This is the one that, of course, gives coronaviruses their name because of the corona-like halo effect seen during a solar eclipse that looks like this or a corona-like appearance of these viruses under the electron microscope.
The spike protein, as we'll talk about in a minute, is critical to the viral entry process. Also, in red is a membrane glycoprotein called matrix protein. This is the most abundant protein on the outside of the viral particle and its function is basically to connect the membrane to the nucleic acid, which is why you can see it in a kind of box there. This is a transmembrane protein, but it has a significant C-terminal domain, which makes contact with the nucleocapsid protein nucleoprotein, and that is probably important for the morphogenesis phase of the viral life cycle, when these virions are formed.
And another minor envelope protein called E is also present. It is also thought to be important for the formation of these viral particles at the end of the viral life cycle. A little more about the spike protein, there have now been a few different research articles published that show structural information of the coronavirus 2 spike protein. And what this, I got it from one of the articles cited below and what This shows here is the structure, a cryo-electron microscopy structure of the coronavirus 2 spike protein superimposed showing the sequence conservation of related spike proteins from other coronaviruses that basically map onto the spike structure of SARS-CoV- 2.
And then they are color coded based on their level of conservation in these related viruses. And what you'll notice in the spike is that this spike is a trimeric protein. What you'll notice is that there are sort of two domains. There is this upper globular domain which is the receptor binding domain. This is what activates the host cell receptor and we'll talk about that on the next slide and then in this domain you'll see that there are many residues that are colored in a sort of bluish green color and this indicates that they are highly variable.
In fact, the receptor binding domain on the spike protein is the most variable part of the coronavirus genome and this tends to be common for viruses in general. This is a region of viruses that is under intense evolutionary pressure due to interactions with the immune system. The bottom part of this spike protein is the part of the protein that encodes and has the fusion machinery that is important for the entry process, and you'll notice that this in purple is much more conserved and is also kind of a classic finding that Fusion machinery tends to be highly conserved.
And hidden in the center of the fusion machinery is actually this hydrophobic fusion peptide, which is very important in being able to fuse the viral membrane with the host membrane so that the virus can deposit its nucleocapsid payload in the cytoplasm of thecells. So what is this entry process like? Well, as I mentioned, the spike protein is the protein responsible for activating a cellular receptor. And can this be imagined as a lock and key mechanism? where the key is the viral glycoprotein and the lock is the cellular receptor. Different viruses will use different cell receptors as a way to enter cells.
The receptor that we know for both SARS-CoV-1 and CoV-2 is the same protein. It is a cellular protein called angiotensin-converting enzyme 2 or ACE2. And that binding to that protein is important, but it is not enough; it needs a second feature to occur and that is a proteolytic cleavage event. And this is done by a cellular protease called TMPRSS2 and perhaps others, but which people have suggested is clearly involved in the entry of coronavirus 2. So what happens is that the spike protein interacts with the receptor. This protease then comes and cleaves the spike protein. There are actually two cleavage events, at least two cleavage events that are known for SARS coronavirus and probably for CoV-2.
In these cleavage events, the first thing that happens is that the receptor binding domain of the spike protein is separating from the fusion domain and the second cleavage event, not shown here, is actually an activating fusion event. that activates the fusogenic state of this protein. And that allows for subsequent entry, which in the case of coronaviruses can occur directly at the plasma membrane, can occur in cytosis, or can occur at both sites. That hasn't really been resolved. So the spike protein is actually a classical class 1 fusion protein and there are several viruses that have fusion proteins of this type.
The best characterized are the flu, the hemagglutinin protein for it. There is an Ebola virus fusion protein that is also class 1. The HIV fusion protein is also class 1. So what I have outlined here at the bottom are the basic steps that are known to underlie the fusion mediated by these class 1 fusion proteins. So, first of all, in the pre-fusion state, you can think of this as almost a metastable state for the fusion protein. And before the proteolytic event that triggers the fusion process, this receptor-binding subunit, which has not yet been cleaved, can basically be thought of as sort of holding down the fusion subunit and keeping it hidden and inactive until the viruses find it. the appropriate place. host cell and can be activated by these proteolytic cleavage events.
So the protease cleavage we talked about causes the receptor binding subunit to move out of the way and that frees up the fusion subunit so that it can then form a pre-hairpin that embeds itself in the target membrane of the cell and This occurs through the fusion peptide. The fusion peptide is a series of hydrophobic amino acids. This usually means that they can be inserted into the membrane. This pre-hairpin then begins to fold back, essentially forming a bundle of six helices and progressively bringing the cellular and viral membranes together to promote fusion. And the final post-fusion conformation in these class 1 fusion proteins is always a hairpin trimer.
And through this mechanism, once fusion has occurred, the viral nucleocapsid with the genome payload can be deposited directly into the cytoplasm of the cell. Some of the early studies that have now emerged of SARS-Cov-2 indicate that there are some interesting features that are different between its spike protein and that of the original SARS-Cov-1. And the first difference is that scientists know, from research with the SARS-CoV-1 spike protein, that there are basically six critical amino acids within the receptor binding domain that are necessary for interaction with the ACE2 receptor and Curiously, five of those six residues. They are different for SARS-CoV-2 than for SARS-Cov-1.
However, CoV-2 can still interact quite efficiently with the ACE2 receptor. The second notable difference is that only SARS-CoV-2 appears to acquire a polybasic cleavage site. This polybasic cleavage site is interesting and important because it is predicted to allow cleavage by other cellular proteases in addition to the one we mentioned. It may also allow efficient cleavage by the cellular protease, TMPRSS 2 protease, known as the canonical type that has been considered for this virus. And it is particularly important because the insertion of a polybasic site in other viruses has been shown to increase transmissibility, particularly for pathogenic influenza viruses.
Therefore, it will be important to determine if the same is true for SARS-CoV-2. Okay, that covers entry and now let's go ahead and talk about what happens to the viral genome once it has entered the cytoplasm of the cell. Well, the 2019 CoV-2 genome has been annotated and depending on which annotation you look at, it is believed to have around 14 open reading frames, encoding approximately 27 proteins. Now let's think about this for a minute because it is something extraordinary. Remember that the viral genome is a single, incredibly long stretch of RNA. It is 30 kilobases long. But for any virus, the same goes for coronaviruses: once RNA is deposited in the cell, the ability to translate or generate proteins from that RNA requires the virus to basically follow the gene expression rules set by the cell.
Guest. And for eukaryotes, translation is a process that is generally monocistronic, meaning that a ribosome arrives and recognizes an RNA in the cell. It will generally translate an open reading frame: a gene from that RNA before it is recycled and falls off. This is different to prokaryotes, which of course have multicistronic RNAs where multiple proteins can be translated from the same RNA, which is generally not true in eukaryotes. So, how is it possible that from one RNA this virus is capable of expressing 27 different proteins? Well, for coronaviruses, there are at least three solutions or three well-known solutions that the virus has developed to solve this problem of expression of many proteins using the eukaryotic rules of gene expression and translation.
And we are going to talk in some detail about them. The first is that, if you look, a large portion of the genome is made up of a single open reading frame, called open reading frame 1, which is separated into sort of two sub-open reading frames, 1a and 1b. a giant open reading frame that basically translates into what's called a polyprotein. It is a series of many proteins fused together without stop codons that intervene to generate a giant protein, which is then processed proteolytically and we will see that in the next slide. This protein is also generated in two different ways by using a programmed ribosome frameshift event, which we will also talk about on the next slide.
That allows it to translate all the proteins encoded in the open reading frame in that portion of the genome, which are generally the non-structural proteins of the genome. But it does not provide the translation of all the structural proteins and other accessory proteins that are found in the 3-major half of the genome, the 3-major end of the genome. And to make them, the virus uses a very unusual strategy of discontinuous transcription that produces something called subgenomic RNA and we will discuss those as well. So to start, remember that the virus first has to make proteins that will be necessary to be able to copy its genome and transcribe the rest of its genes and, for any RNA virus, this requires an RNA-dependent RNA polymerase or an RdRP.
If you are a positive sense RNA virus, like coronaviruses, your incoming genome is basically recognized as a messenger RNA, it is ready for the ribosomes. So you don't need to package that RNA-dependent RNA polymerase complex or protein into your virion because it can be translated directly from the genome and that's what happens. That's what this gigantic open reading frame 1a or 1ab encodes. So, as I mentioned, this becomes a huge polyprotein. Within this polyprotein are two proteases that the virus encodes and these proteases, the job of those proteases now is to basically break down this giant polyprotein as shown here on the left in the bottom box into individual proteins that will have separate functions for the virus. genetic expression and replication, so proteolysis is obtained to generate many different proteins from an initially translated polyprotein.
Also, you'll notice that this polyprotein, as I mentioned, doesn't just translate as a giant open reading frame to begin with. There is a frame change event. So some of the time, maybe 50 or 60 percent of the time, the ribosome will read and there will be a stop codon at the end of ORF 1a, so it will stop there. However, the remaining percentage of the time viruses allow the ribosome to read that stop codon and continue translating it to generate a longer ORF 1ab fusion. And that programmed translation is a frameshift event that is governed by two properties of genomic RNA.
The first is that right around that stop codon, there is something called a slippery sequence and this is shown in the RNA diagram on the right, the sequence of UUUAAAAC and when the ribosome lands on this site it is known that it tends to have a propensity to occasionally goes out of frame. Now, the frequency with which that frameshift event occurs can and does increase in these coronaviruses because right after that slippery sequence is what's called an RNA pseudoknot structure. This is basically a highly stable RNA structure that causes the ribosome to stop when it encounters it, so the structure is thought to interact with the ribosome, causing the ribosome to stop on the slippery sequence, increasing the chances of it going out of frame again. if you move one nucleotide back out of frame, that stop codon at the end of ORF 1a is no longer read as a stop codon and the ribosome can continue translating and generating the rest of the viral polyprotein.
Okay, so that protein is processed as I mentioned, but how do you get the production of the rest of the proteins that are at the 3 end of the viral genome? Structural and accessory proteins. These are basically made of a nested set of what are called subgenomic RNAs that all have 3-prime coterminals. So this is important if you think about how they are going to express their proteins. These are not polyproteins, but by having this nested set of subgenomic RNAs, what it allows the virus to do is have each of these genes at the 3-major end of the genome, have the opportunity to be present as the 5-major end more open reading frame in a messenger RNA.
Let's think about it this way: if you are generating an RNA, for example, where in this case gene 2, which would represent the spike protein, for example, is at the leading 5 end, the ribosomes are going to translate gene 2. and everything What lies behind it, based on eukaryotic translation rules, will basically be seen as a UTR sequence, an untranslated sequence. So in this case only gene 2 will be translated into protein 2 or spike. The same thing if you generate a transcript where now gene 3 has the possibility of being the top 5 open reading frame that will be translated into protein and everything downstream will be an untranslated sequence.
So each gene at the top 3 end of the viral genome has the potential to be the top 5 open reading frame in a messenger RNA, allowing it to be translated. How this happens is quite fascinating and involves another feature, which I hope you have noticed here in these RNAs that I have drawn and that is that in addition to being coterminal 3 primes, they all have exactly the same sequence at the 5-. leading end and that same exact sequence is the same sequence at the 5-prime end of the genomic RNA called the leader or L sequence. So how is it possible to get the same sequence, which is not present at this triple end of the genome?
How can it be fused to the ends of each of these subgenomic RNAs? And basically the answer to that lies in how they are produced. It involves a series of sequences called transcription regulatory sequences or TRS. At the junction between each of those virus-encoded genes, as well as at the 5-core end of the genomic RNA just downstream of the leader sequence, indicated here in red, are these conserved TRS sequences, these regulatory sequences. transcriptional. And so, as the polymerase comes and copies the genome, it will come to these TRS, which are at the leading 5 end of each of the genes.
There is a highly conserved core sequence within these TRS. This is called the core sequence or denoted as CS here in yellow. And so once the polymerase gets to these TRS and copies this core sequence, it can continue copying or it will now jump from that sequence, probably through a long-range RNA-RNA interaction and a base pair with the same core sequence. which is part of the TRS at the 5-prime end of the genomic RNA which is just downstream of the leader and then the polymerase will continue to transcribethere capturing the leading sequence. This looks like this, where the nascent RNA is shown in red.
RNA polymerase begins to copy itself. You will see that the TRS are present just before each of the virus genes. As the polymerase gets to one of the TRS, it can either read that TRS and move on to the next one or it will jump and basically move to the TRS at the leading 5 end of the genome and finish its transcription to generate that fusion. with the leading sequence. So this is a discontinuous transcript. It then allows the generation of a series of these subgenomic templates. Remember, these are copied from the positive sense RNA genome. So these are now negative sense or negative sense RNA.
They are complements to the genome but are not prepared for ribosomes. To do that, the polymerase now has to re-make copies of these negative-sense subgenomic messenger RNA templates to generate the actual positive-sense messenger RNAs that can be translated. It is worth thinking about that this discontinuous transcription mechanism means that there are many polymerase jumps and probably facilitates what are known to be extraordinarily high recombination rates within coronaviruses. As far as I've heard estimates are around 25%. Most RNA viruses and positive-sense RNA viruses have extremely low levels of recombination, so this is a unique feature of coronaviruses that may be interesting as to how they evolved.
And perhaps also how they are able to maintain such huge genomes. So this discontinuous transcription mechanism is quite complex and is orchestrated by a replicase that includes the polymerase, but also many other proteins. And this replicase complex requires functional integration of RNA polymerase, capping and proofreading activities, among other things. And so what I'm showing you here on the left is a structure of basically what people think is the polymerase holoenzyme, which is made up of the RNA-dependent RNA polymerase nsp12 along with two other nonstructural proteins, nsp7 and nsp8. , which is believed to help with the processivity of the RdRP.
As I mentioned, this is thought to be perhaps the core holoenzyme of the polymerase and is thought to be capable of initiating de novo primer-independent RNA synthesis. Furthermore, the complex associates through protein-protein interactions with another non-structural protein called Nsp14, which is a bifunctional protein that has capping activities and exonuclease activity, which turns out to be a real activity that changes the paradigm of how scientists They think about RNA. evolution of the virus. I'm going to spend some time talking about that, but first I'll mention that it's not just these, but the proteins mentioned above, but in fact, there are a variety of other viral processing proteins and activities associated with the complex. replicase, not all of which are well understood biochemically, as well as an indefinite set where at least one incompletely defined set of cellular proteins may also participate in their regulation.
So there is a very complicated replicase complex involved in orchestrating this discontinuous transcription mechanism. Well, let's go back to this exonuclease that I mentioned as part of the polymerase complex. It turns out that the theoretical limit of how large an RNA virus's genome can be is about 30 kilobases. And this theoretical limit comes from the observation that in RNA viruses, all of which have RNA-dependent RNA polymerases, these RdRPs do not have proofreading capabilities. This is different from the polymerases in our own cells. And this means that they are error-prone, and this error-prone ability of RdRPs underlies the massive evolution that occurs during the replication of many RNA viruses to generate things called quasispecies and swarms of mutants that are very characteristic of infections like HIV and the flu.
And what it also means is that most viruses actually don't even come close to that theoretical 30 kilobase limit. Most RNA viruses are well under 20 kilobases and most are in the order of, you know, 10 to 12 kilobases, maybe. Now, there are viruses like coronaviruses, as I mentioned, and others in a larger group of similar viruses called Nidovirales that have surprisingly large RNA genomes: 30 kilobases. We even know of some that now exceed 30 kilobases. Even thus exceeding the theoretical threshold. And within these viruses, only these viruses, not all but many, have this exonuclease activity present, and this led to the idea that this exonuclease activity might actually be conferring a proofreading function to the RdRP, which as I said What was mentioned was a real paradigm shift in thinking about how RNA-dependent RNA polymerases might actually be able to correct.
So, in fact, in the SARS coronavirus, if the ExoN, this exonuclease gene, is mutated and then you measure the number of substitutions or mutations that occur during the replication of this virus, you can see here in this graph that, Compared to the number of mutations that occur in the wild-type virus, there is a full jump of more than 20 in mutational frequency in the virus lacking this ExoN activity. So here you can see this spread through the rest of the coronavirus genome,
focus
ing first on the top panel. Where in dark black are basically the lines that show the mutation frequency in the populations during infection with a wild type virus and the gray lines show the same during infection with the ExoN mutant virus, and you can see that there is an increase significant in number and distribution of mutations that are acquired.This also causes these viruses to have an ExoN mutation and makes them hypersusceptible to mutagens, as shown in the bottom panel, which includes here what was tested is 5-fluorouracil, so you can see, of course, the Treatment with 5-FU, which is a mutagen, increases the mutational frequency of the wild-type virus, but it further increases the mutational frequency, of course, of this ExoN mutant. So it's also interesting that you would expect that if this exonuclease activity was what allows viruses to reach these enormous genome lengths, it would be absolutely essential for the virus and for some viruses as they are.
They cannot tolerate a mutation in the ExoN, but the SARS coronavirus and some others, in fact, although they are attenuated mutants, can evolve and adapt over multiple passages to stabilize populations and, in fact, prevent lethal mutagenesis . So the location of these will be what you might think of as sort of suppressor mutations in the genome, you would expect them to do things like increase the processivity, perhaps of RNA-dependent RNA polymerase, and they may be doing other things as well. things. So I think it's a really interesting concept to think about and, in fact, in the murine betacoronavirus called MHV, an ExoN mutant showed clear promise as a vaccine strategy at least when it was used in mice because it was an attenuated strain, but subsequently allowed protection against a challenge with a wild-type strain.
This nsp14, which is the exonuclease, is really a fascinating protein. It is a bimodular protein that is composed of two different domains that basically have two different activities. Here is this ExoN domain, which is involved in proofreading and then there is also a domain that is a methyltransferase domain that is thought to be involved in the messenger RNA limitation reaction. And these two domains are separated by a flexible hinge region and probably allows them to orient the protein in different ways as these different functions are needed. And ExoN works in conjunction with another non-structural protein called nsp10.
Together they function as a heterodimer and basically function in a mismatch repair mechanism. So actually ExoN, this proofreading activity can efficiently remove ribavirin, which is a chain terminator that is commonly used as an antiviral against many different RNA viruses. But it's known that it doesn't work against coronaviruses, and that's because this proofreading activity can basically remove that nucleoside analog and allow the virus to continue to replicate. It has been shown with this mouse coronavirus, MHV, that a knockout of ExoN1 is inhibited more efficiently than the wild-type virus. -Remdesivir-like virus, which is another nucleoside analogue that is being widely explored at the moment for its potential to block CoV-2 replication.
And what that suggests is that ExoN probably also reduces the uptake of Remdesivir. For that reason, it is probably beneficial to try to simultaneously target both RdRP with Remdesivir and ExoN with some type of specific exoribonuclease inhibitor. Alright, now that I've explained how the virus is able to replicate its genome and cause its genes to be expressed through this incredibly sophisticated replicase complex, I'm going to move on to talk about where this happens in a cell because it turns out that the virus is able to form these very intricate membrane structures called replication and transcription complexes.
So these are basically interconnected double-membrane vesicles where viral replication and transcription can occur. And I show you here some images from a reference that I cited below and that come from a cryo-electron tomography of cells infected with coronavirus. And you can see on the far left an EM image showing one of these classic double membrane vesicles that form in infected cells. And in the center is a further image, where you can see that the cell basically now contains many of these double membrane vesicles and, on the far right, what you see is a representation of the 3D surface of a chamber cryogenic. -Electronic tomography of these, where you can see that the interior of these membranes is shown in violet color.
And many of them are actually interconnected in the sense that the outer membrane encapsulates several of these vesicles at once. These convoluted membranes are derived from the endoplasmic reticulum and, as I mentioned, many of the double-membrane vesicles seen in these tomography experiments are actually interconnected by their outer membrane and are part of an elaborate network that is contiguous to the rough endoplasmic reticulum. . . Within these compartments is where viral replication and transcription is believed to occur. And this works for the virus and probably benefits it in multiple ways. Firstly, by compartmentalizing them they can protect their genome from possible attacks by antiviral mechanisms or other exonucleases or nucleases that could be generally present in the cytoplasm.
It can also help them concentrate the factors necessary to efficiently replicate and transcribe the viral genome. Because these replication compartments, these are RTCs, are essential for virus replication, they have been discussed as potential antiviral targets by attempting to disrupt the formation of this membrane. A lot of work has been done to try to explore how they form. And what is known is that there are integral membrane proteins that are part of the replicase complex and that are believed to function in vesicle biogenesis. And the three replicase components that are predicted to at least have transmembrane domains are nsp3, 4, and 6.
And they are thought to be directly involved in vesicle formation. In a study that I cite below, it has been shown that two of these nsp4 and nsp3, when expressed alone outside the context of infection, are actually sufficient to drive the formation of these double membrane vesicles and it is believed that this occurs by an interaction between the luminal loops of these proteins that drives membrane curvature and vesicle formation. So recently there has also been work to try to identify which proteome components are basically associated with these replication and transcription complexes. And this has been studied with the mouse coronavirus, MHV, using a proximity labeling-based approach involving the biotin ligase BirA that was fused in the context of the virus with one of these replicase proteins nsp2, which is known to localize within these replication compartments and therefore through the addition of biotin which could then be transferred to the proximal proteins, these proteins could then be purified, identified by mass spectrometry to basically identify the RTC proteome and then, in this particular study that I cited below, then they took these hits and did a targeted siRNA screen to determine which of the components that are host factors are actually necessary for viral replication, which are the proviral factors here.
And I want to point out that they ruled out attacks that compromised cell viability on their own, so these are attacks that decrease coronavirus replication, but do not affect cell viability. And what they noticed, of course, is that there are a number of things involved in cellular transport that are not and vesicle formation that would be expected and our results interesting for future follow-up, as well as a number of catabolic processes.This finding is quite interesting, as it could provide a link to the described coronavirus transcription and replication complex, which encodes the nsp3 protein, which is believed to have deubiquitination activity.
And then, interestingly, some of the major successes were in the translation machinery, these eIF3 components of the translation complex. And they were able to use fluorescence imaging of cells labeled with pure myosin, which is basically a form of pulse labeling, to detect nascent transcription. And this showed a really pronounced enrichment of actively translating ribosomes near these viral transcription and replication complexes, particularly in early to mid infection, indicating that the translation machinery, in addition to the transcription machinery, is basically recruited near these membranous networks. Furthermore, it was interesting to observe which viral proteins are present within these membrane complexes.
And in pink are the viral proteins that were significantly enriched and it makes a lot of sense because most of them are non-structural proteins that are known to be involved in replication and transcription, so they should be there. It's also interesting to note what wasn't there. And so, for example, one of the proteins that was not significantly enriched is a non-structural protein called nsp1. Nsp1 is fascinating. It is a key pathogenicity factor of the coronavirus. It is a host shutdown factor that basically restricts gene expression coming from the host cell and it does so through a two-pronged approach.
Nsp1 is able to directly interact with the 40S subunit of the ribosome and in this way block the translation of host RNAs and also mediate the endonucleolytic cleavage of these RNAs in a rather generalized manner, leading to extensive and accelerated RNA degradation. messenger in these cells. And this benefits the virus, perhaps for at least two reasons. A classic way to think about why host shutdown benefits the virus is that it helps viruses divert the host cell's gene expression machinery toward viral needs. The second reason, which has been directly demonstrated for these coronavirus nsp1 proteins, is that this is a general immune evasion tactic because, by promoting widespread RNA degradation, many of these RNAs will be induced as part of the interferon response. and this helps the virus delay the response to interferon.
In particular, for nsp1, it appears to be specific for cleaving host RNAs because the leader sequence, the top-5 leader sequence that we talk about for subgenomic RNA synthesis and which is present in genomic RNA, appears to protect viral transcripts. of nsp1-mediated cleavage. . And this is a selective shutdown of the host, but not of the viral RNA. This particular activity, the idea that it blocks the interferon response, is quite relevant to viral pathogenesis. In fact, it has been shown that if this nsp1 protein is mutated, and this is a survival curve of a mouse, you can see that while mice infected with a wild-type virus generally die about 6 days after infection .
In the absence of this key virulence factor, all mice survive infection. Therefore, this mutation of this factor has also been something that has been explored as a possible vaccine strategy. All right, beyond that nsp1 virulence factor, several of the other things that are not present in these RTC complexes, if you look at them, are basically virion and assembly proteins, things like the matrix protein, the envelope protein, the spike protein and that makes a lot of sense because viral morphogenesis or assembly does not take place in these RTCs. This is happening in some sort of presumably discrete location, so it makes sense that they're not part of these RTC complexes.
Furthermore, many accessory proteins are not part of the RTC complexes. And then what are accessory proteins? These proteins and genes are elements of viruses in general that tend to be specific to a particular viral species or a particular viral genus. And accessory genes are often dispensable for viral replication in tissue culture cells, but they play really important roles in virus-host interaction in an in vivo context with the animal or human. So what I'm showing you here in this diagram are the accessory proteins, which are labeled in blue for several representative betacoronaviruses. You can see that SARS-CoV-2 is included in this diagram in the center, in direct comparison to SARS Coronavirus.
You can see that they do in fact share a number of accessory proteins that look quite similar, but I think it will be interesting to compare the differences in the future as there are, at least from sequence observation, several notable SARSs. Coronavirus 2 variations in these accessory proteins. In particular, in accessory proteins that participate in the interaction with the innate immune response and perhaps counteract the response to interferon and some of them are listed here in this table, accessory proteins 3a, b, open reading frame 6, reading frame open 8. Each of these has some notable differences. You'll also notice that I'm not going to go over the functions of all those on the table, although I will point out that even for SARS coronavirus and other coronaviruses the functions of many of the accessory proteins are only partially performed. was or not yet established.
This will be an important area for future research. Well, we've talked about the composition of these transcription and replication complexes that form from this elaborate network of ER-derived vesicles. And then once the viral genomes replicate within these, they need to assemble into new viral particles, and this is called viral morphogenesis. And assembly is basically driven first by the association of the nucleocapsid protein with the genomic RNA. This is assembled to form these helical nucleocapsids, remember that they are formed in the center of the viral particle. These then need to associate with the components of the viral membrane.
And these are the spike protein, the matrix protein, the envelope protein. These are all integral membrane proteins that insert into the endoplasmic reticulum, and then the nucleocapsid, which is attached to the viral genome, then buds into these. Perhaps in the intermediate Golgi compartment of the ER or labeled here as ERGIC, budding is known to occur in association with the Golgi and then these particles are probably glycosylated at particular sites and released through an exocytosis-like process, outside of the cells so that they can then continue and infect neighboring cells. Well, those are the basic mechanisms of virus replication and now, in the last few minutes, I want to move on to the immune interactions of this virus.
First I want to point out that SARS and MERS coronaviruses and we still don't know the answer for CoV-2, an interesting feature of these viruses is that they induce very little or no interferon in most cells. And this is illustrated in this image that I have shown here, where you can see that the top gel shows a signal for interferon beta and in the control cells and these are infected with Bunyavirus, which is a negative sense RNA virus, which it clearly induces interferon beta quite strongly as many RNA viruses do. The SARS coronavirus contrasts sharply with that control and very little induced interferon beta signal can be seen.
And then why is it like this? Well, we've already touched on this a little bit, but just to emphasize that there are a number of putative interferon antagonists that have been identified in the SARS coronavirus genome, several non-structural proteins that we talked about nsp1, several accessory factors that I also mentioned that the matrix nucleoprotein can also counteract this. So it appears that this virus and perhaps also CoV-2 actually have a multi-pronged approach to dampening the early interferon response to the virus. And this is thought to be really important for viral pathogenesis. And in fact, the pathogenesis of SARS has been shown to be related to delayed interferon 1 signaling and subsequent immune toxicity, so let's look at this first in terms of this survival.
Shown here is a graph for mouse experiments where you can see that wild-type BALB/c mice, when infected with coronaviruses, tend to succumb to the infection approximately 6 to 8 days after infection. However, if you infect mice that lack interferon signaling, so they lack the Ifnar receptor, knock it out for that, you infect these mice with a wild-type virus and none of these mice die. This suggests that the response to interferon is ultimately related to the death of these mice after coronavirus infection. And that's not because these mice that lack the Ifnar protein can replicate the virus differently and that's shown here in this graph in panel D, where you can see that the replication is the levels of virus replication, measured by plate-forming units. in the lung are basically very similar between wild-type mice and Ifnar knockout mice.
So the hypothesis is that the virus is able to replicate initial titers that are too high because of these accessory factors and other multiple approaches that it has to delay the response to interferon. But then an interferon response appears later and at an inappropriate time because it can no longer be used to stop the initial viral infection, but what this response is doing is driving the aberrant recruitment of pathogenic inflammatory monocyte macrophages and activation of the immune system innate. response leading to cytotoxicity. And that's shown here in these diagrams, where on the left you have an uninfected alveolus and these cells, upon acute coronavirus infection, begin to engage in rapid replication of the virus because the virus is impeding the antiviral response to early interferon.
This leads to the infiltration and release of inflammatory cells, probably from both the infected cells and these infiltrating inflammatory cells, proinflammatory cytokine and chemokine responses, and it is these immune responses that are thought to lead to acute lung injury and death. acute respiratory distress syndrome. Therefore, there is a clear immunopathology associated with these infections. Finally, I want to point out that it has been shown for SARS and also for circulating human coronaviruses that neutralizing antibody titers, shown in the graph here, and memory B cell responses, not shown here, are Of short duration. Patients recovered from SARS.
So the black line here shows a cohort of SARS patients who were monitored for neutralizing antibodies and can be seen to generate a robust neutralizing antibody response; However, this response is not sustained and a couple of years after the initial infection their response is basically disappearing. A couple of atypical patients are now seen shown in the green line and the orange line, indicating that some people can mount a sustained protective response, but for most people infected with the virus immunity probably wanes and I think that is going to increase. It will be important to think, in particular, whether that is also the case for CoV-2 and what that means for the continued circulation of this virus.
So I think there are a number of really important immunological questions that need to be answered for CoV-2 right now that will really greatly inform how this virus causes the pathogenesis and control of the pandemic. And I just summarized some of these here. For example, what does seroconversion for CoV-2 look like? How long do recovered people remain immune? And can they be reinfected? What kind of immunity will we get from vaccines? And how does it compare to the response to infection, which I have shown here? We also need more information about what is happening in the older population, particularly as it relates to their immune responses, immunology, and the inflammation that occurs in these patients.
Because in part this will help scientists identify parallels that should be looked for in animal models and these animal models themselves need significant development for CoV-2. Well, with that I just want to finish by listing some of what I think are some of the key open basic scientific questions about these viruses. First of all, for SARS-CoV-2, what is the role of the polybasic view on the spike protein in the transmission of CoV-2? Is this really a component that has helped accelerate the transmission of this virus? What are the pathways involved in coronavirus-induced membrane remodeling and how do replication and transcription complexes temporally and functionally coordinate the different stages of the viral life cycle?
What are the biochemical activities and functions of the various proteins that make up this highly sophisticated transcription and replication complex? How do replication and transcription coordinate indifferent stages of the viral life cycle? How do these coronaviruses maintain such a large genome and still have sufficient mutation rates for adaptation and movement between species, which we know certainly occurs with these viruses? What are the functions of CoV-2 accessory proteins and how do they affect in vivo growth and virulence of the virus? And will people infected with coronavirus 2 or vaccines generate long-term protective immune responses? Well, I'm going to end this and first I want to acknowledge that I received a lot of help in collecting information and slides for this talk from Professor Laurent Coscoy, as well as members of my lab: Divya Nandakumar, Ella Hartenian. and Michael Ly, Azra Lari, Jessica Tuckers, and Allison Didychuk.
I'd also like to mention that if you're not a virologist but are curious about how viruses and viral research have really contributed greatly to the basic understanding of molecular biology, I've recorded an open access iBio talk about that. which link is below and most importantly I really want to thank all the coronavirus researchers who generated all the data sets that I talked about today and who are also playing really key roles in the response to this current pandemic. like all the scientists and medical staff who are working tirelessly to fight the pandemic, and we are deeply indebted to them.
So with that, thank you very much.
If you have any copyright issue, please Contact